APRANK: computational prioritization of antigenic proteins and peptides from complete pathogen proteomes
Abstract
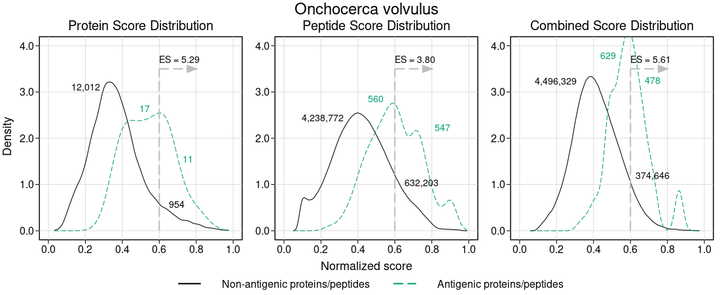
Availability of highly parallelized immunoassays has renewed interest in the discovery of serology biomarkers for infectious diseases. Protein and peptide microarrays now provide a rapid, high-throughput platform for immunological testing and validation of potential antigens and B-cell epitopes. However, there is still a need for tools to prioritize and select relevant probes when designing these arrays. In this work we describe a computational method called APRANK (Antigenic Protein and Peptide Ranker) which integrates multiple molecular features to prioritize antigenic targets in a given pathogen proteome. These features include subcellular localization, presence of repetitive motifs, natively disordered regions, secondary structure, transmembrane spans and predicted interaction with the immune system. We applied this method to prioritize potentially antigenic proteins and peptides in a number of bacteria and protozoa causing human diseases: Borrelia burgdorferi (Lyme disease), Brucella melitensis (Brucellosis), Coxiella burnetii (Q fever), Escherichia coli (Gastroenteritis), Francisella tularensis (Tularemia), Leishmania braziliensis (Leishmaniasis), Leptospira interrogans (Leptospirosis), Mycobacterium leprae (Leprae), Mycobacterium tuberculosis (Tuberculosis), Plasmodium falciparum (Malaria), Porphyromonas gingivalis (Periodontal disease), Staphylococcus aureus (Bacteremia), Streptococcus pyogenes (Group A Streptococcal infections), Toxoplasma gondii (Toxoplasmosis) and Trypanosoma cruzi (Chagas Disease). We have tested this integrative method using non-parametric ROC-curves and made an unbiased validation using an independent data set. We found that APRANK is successful in predicting antigenicity for all pathogen species tested, facilitating the production of antigen-enriched protein subsets. We make APRANK available to facilitate the identification of novel diagnostic antigens in infectious diseases.
Alejandro Ricci
Former Lab Member (2016 – 2023)
PhD Fellow. Processing big data to draw biological conclusions.
Santiago J Carmona
Former Lab Member (2007 – 2015)
Present address: Ludwig Center for Cancer Research, University of Lausanne
Fernán Agüero
Principal Investigator, Assistant Professor
Using and generating data to guide discovery of new drugs and diagnostics.